Tables with lots of text in LaTeX often lead to tables that do not fit on a page. This post shows how to produce tables with automatic line breaks. Here is a simple example of a badly formatted table:

The tabularx package has the possibility to break lines automatically by using the column specifier X:
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 |
\begin{table}[!htbp] \centering \begin{tabularx}{\textwidth}{lX} \toprule Number & Description \\ \midrule 1 & This is a mighty long text that could go over multiple lines but in a normal table is shown on one line and therefore the table does not fit on the page \\ 2 & This is a mighty long text that could go over multiple lines but in a normal table is shown on one line and therefore the table does not fit on the page \\ 3 & This is a mighty long text that could go over multiple lines but in a normal table is shown on one line and therefore the table does not fit on the page \\ \bottomrule \end{tabularx}% \label{tab:addlabel}% \caption{A table with line breaks} \end{table}% |
This results in the following table:

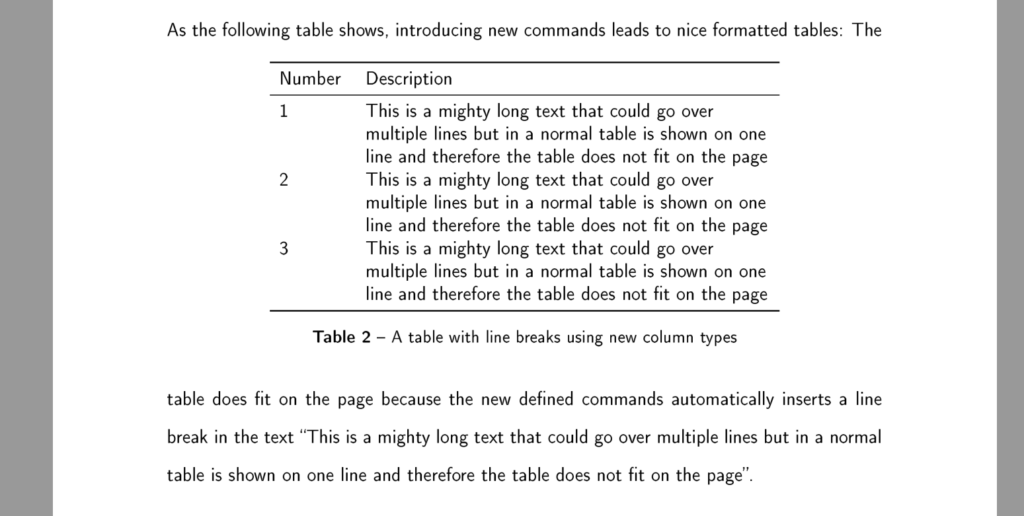
Another option is defining a new command to let you set the width of the columns:
|
1 2 3 4 5 6 7 8 |
% left fixed width: \newcolumntype{L}[1]{>{\raggedright\arraybackslash}p{#1}} % center fixed width: \newcolumntype{C}[1]{>{\centering\arraybackslash}p{#1}} % flush right fixed width: \newcolumntype{R}[1]{>{\raggedleft\arraybackslash}p{#1}} |
This new type has the nice feature that you can define the width of the columns yourself. It can be used as follows:
|
1 2 3 4 |
\begin{table}[!htbp] \centering \begin{tabular}{L{1cm}L{9cm}} ... |
with the following result: